
IVDR – What it is and why early compliance matters
Practical Implications for Manufacturers – Conformance with the IVDR is a significant business challenge that must be overcome – don’t be late!

In order to enjoy continued access to the EU Market – medical device manufacturers must be proactive and begin preparing now.
The European Union Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) came into effect as of May 26th, 2017. The clock is ticking for current Medical Device manufacturers in the European marketplace to develop and implement strategies to be fully compliant.
What is the In Vitro Diagnostic Regulation (IVDR)?
The IVDR is the new regulatory basis for placing on the market, making available and putting into service in vitro diagnostic medical devices on the European market. It will replace the EU’s current Directive on in vitro diagnostic medical devices (98/79/EC).
As a European regulation, it will be effective in all EU member states and EFTA states immediately without need to be transferred into the law of respective states, however national laws may be adapted to back up some requirements in more detail.
Increased regulatory oversight, expanded clinical evidence requirements, greater requirements for IVD manufacturers and intensified supervision of Notified Bodies are just some of the major changes to be considered, as compared to the EU’s previous Directives: the Active Implantable Medical Devices Directive (AIMDD), Medical Device Directive (MDD), and In Vitro Diagnostic Directive (IVDD).
These lengthy documents can be cumbersome to navigate, particularly when trying to understand how the new regulations compare to the previous directives—and more importantly, the critical steps that manufacturers should undertake now to ensure conformity by the deadlines.
Advantu, in an effort to assist global medical device and IVD manufacturers with their compliance planning, has made available our process consultants to answer your questions and help you prepare for implementation of the new regulations.
When is the IVDR expected to be implemented?
Manufacturers should undertake steps now to ensure conformity by May 2020 (MDR) and May 2022 (IVDR), respectively.
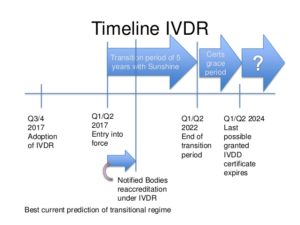
When do medical device manufacturers need to comply with the new IVDR?
Manufacturers of currently approved in vitro diagnostic medical devices will have a transition time of five years, up to 26 May 2022 to meet the requirements of the IVDR.
Products already certified by a Notified Body may be placed on the market for further 2 years under some conditions, e.g. the certificate issued under the IVDD is still valid and subject to surveillance by the Notified Body who had issued it and no significant changes to the product are made.
What are the key changes expected in the new IVDR?
The actual terms of the proposed regulation are subject to change until final publication of the IVDR in the Official Journal of the European Union.
Some of the key changes expected include:
- Product scope expansion. Expanded scope will cover diagnostic (including Internet-based) services, genetic testing and other tests that provide information about a patient’s predisposition to a specific disease or susceptibility for a medical treatment.
- Reclassification of devices according to risk. Risk classes will range from Class A for low risk devices to Class D for high risk devices.
- More rigorous clinical evidence. Manufacturers will need to conduct clinical performance studies and provide evidence of safety and performance according to a device’s assigned risk class.
- Self testing and near patient testing devices will be subject to a premarket approval approach.
- More stringent documentation.
- Identification of ‘person responsible for regulatory compliance’
- Implementation of unique device identification for better traceability and recall
- Requirements for post market surveillance will be reasonably increased and general timeline for reporting reduced.
- More rigorous surveillance by Notified Bodies to reduce risks from unsafe devices
- Greater Scrutiny of Notified Bodies
- No “grandfathering” provisions. All currently approved in vitro diagnostic devices must be recertified in accordance with the new requirements.

What are the implications of the new IVDR for in vitro medical device manufacturers?
The complex development process for in vitro diagnostic medical devices, combined with the anticipated changes, are likely to make the transition a complicated and time consuming process for most device manufacturers.
Manufacturers of in vitro diagnostic medical devices are well-advised to stay current on amendments to IVDR by Implementing and Delegated Acts, as well as additional changes that may impact them.
Advantu’s Consultants help medical device manufacturers in the EU Market better understand the immediate IVDR implications by:
- Analyzing the most influential changes and highlighting their implications for regulated medical device companies.
- Providing practical steps that medical device manufacturers should take as soon as possible to prepare for this substantial change in the European regulatory environment.
How can Advantu Help?
Advantu’s global regulatory experts welcome the opportunity to discuss strategic options related to the new EU MDR and IVDR regulations, including the critical steps you should be taking now to realize successful outcomes.
Please contact us to schedule a Free Consultation so you learn how to plan, and implement the IVDR changes necessary to ensure compliance with the EU’s new Medical Device regulations.


